This site is intended for healthcare
professionals in Belgium and Luxembourg.
CHARACTERISTICS
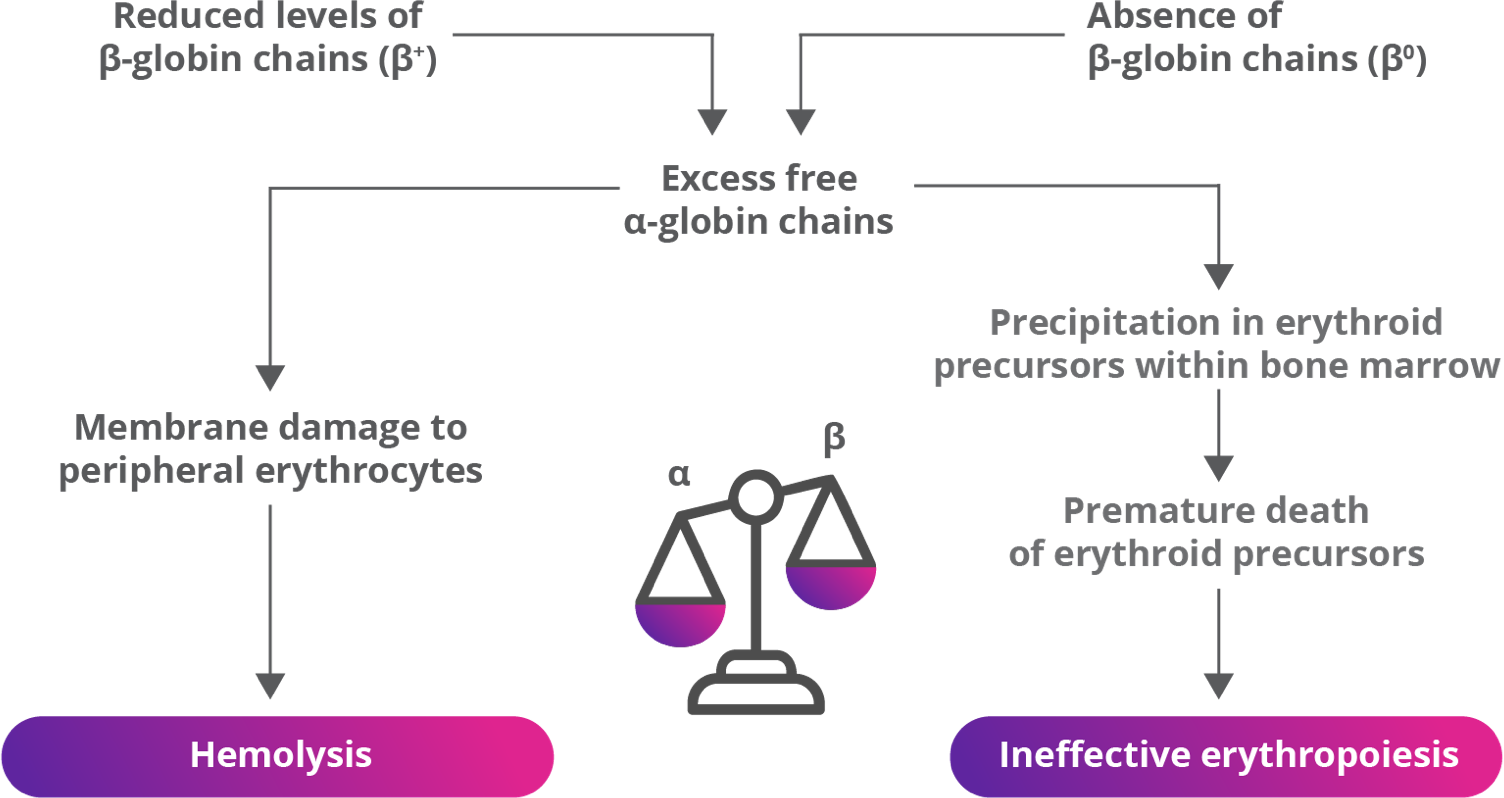
Ineffective erythropoiesis due to mutations in the β-globin gene and hemolysis are hallmarks of
β-thalassemia.1-3
β-thalassemia is a rare genetic condition that reduces production of hemoglobin (Hb). It is characterized by reduced or absent synthesis of the β-globin chain of Hb, decreased Hb in the blood, reduced RBC production and anemia.2,4,5
β-globin gene defects lead to reduced (β+) or absent (β0) synthesis of the β chains of hemoglobin and affected patients can have various combinations of normal (β), β+, and/or β0 alleles.2,4,5
β-thalassemia is caused by mutations in the β-globin gene,
resulting in ineffective erythropoiesis2,4,5
PREVALENCE
An estimated 80 million to 90 million people—approximately 1.5% of the global population—are carriers of a β-thalassemia mutation.6
- β-thalassemia is common in low- and middle-income countries, where malaria remains or was endemic, but with population migration, it has spread to many more regions.6 Evidence suggests that carriers are more resistant to severe forms of malaria, thus they live longer and have more affected children7
- The Southeast Asia region alone accounts for approximately 50% of the world’s carriers, around 40 million people6
- Due to migration from countries with a high prevalence of β-thalassemia, the disease has spread to other parts of the world6
β-thalassemia is most prevalent in the tropics and subtropical regions (see highlighted areas)6,7

CLASSIFYING
β-THALASSEMIA
Approximately 200 disease-causing mutations in the β-globin gene have been documented.6 Traditionally, β-thalassemia was classified into 3 main subgroups8,9:
- Thalassemia major: severe anemia from birth
- Thalassemia intermedia: mild to moderate anemia
- Thalassemia minor: may be borderline symptomatic
Patients are classified as either transfusion dependent or non-transfusion dependent based on the need for transfusions in order to survive5,9
LIMITATIONS OF THERAPY
Current therapy of β-thalassemia is predominantly limited to lifelong red blood cell (RBC) transfusions plus iron chelation therapy (ICT).2,11,12
- RBC transfusions are a life-saving, supportive-care option in transfusion-dependent β-thalassemia, which has reduced morbidity and improved life expectancy of patients11,12
- RBC transfusions rapidly improve hemoglobin (Hb) levels to compensate for anemia; however, they suppress and lack the ability to correct the underlying ineffective erythropoiesis2,11,12
- The improvement in Hb levels is also temporary, hence regular transfusions are needed in transfusion-dependent β–thalassemia11
- Transfusions are time consuming, taking an average of 3 to 4 hours for treatment, including about 1.9 hours on average per blood unit transfused5,13
- There is a risk of infection from RBC transfusions like hepatitis B and C and, in some populations, HIV.14 Alloimmunization can occur in 10%-20% of patients with β-thalassemia5,16
- Iron overload is the greatest concern associated with regular RBC transfusions4,14
- Because there is no physiological mechanism to excrete excess iron, without chelation, iron overload leads to organ failure and mortality in β-thalassemia5,15
ICT is used for the management of iron overload, which is an inevitable complication of regular blood transfusions because the body lacks a mechanism to excrete excess iron.5
- Iron chelating agents can have various side effects, such as retinal toxicity, hearing loss, skin rashes, diarrhea, increased liver enzymes, reduced kidney function, arthropathy, or neutropenia2,15
- With the availability of new ICTs and new formulations, adherence has improved but is not yet optima12,5
Red blood cell (RBC) transfusions are an important supportive-care option; however, they are burdensome and carry risks2,5,13-15

Due to iron overload, patients with transfusion-dependent
β-thalassemia need to receive iron chelation therapy (ICT)5


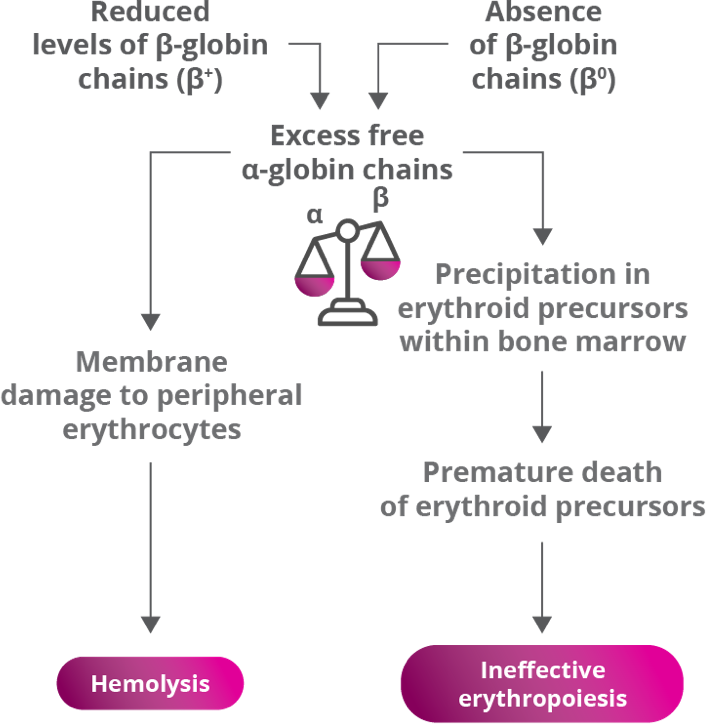
Ineffective erythropoiesis due to mutations in the β-globin gene and hemolysis are hallmarks of β-thalassemia.1-3
β-thalassemia is a rare genetic condition that reduces production of hemoglobin (Hb). It is characterized by reduced or absent synthesis of the β-globin chain of Hb, decreased Hb in the blood, reduced RBC production and anemia.2,4,5
β-globin gene defects lead to reduced (β+) or absent (β0) synthesis of the β chains of hemoglobin and affected patients can have various combinations of normal (β), β+, and/or β0 alleles.2,4,5
β-thalassemia is caused by mutations in the β-globin gene, resulting in ineffective erythropoiesis2,4,5
An estimated 80 million to 90 million people—approximately 1.5% of the global population—are carriers of a β-thalassemia mutation.6
- β-thalassemia is common in low- and middle-income countries, where malaria remains or was endemic but with population migration, it has spread to many more regions.6 Evidence suggests that carriers are more resistant to severe forms of malaria, thus they live longer and have more affected children7
- The Southeast Asia region alone accounts for approximately 50% of the world’s carriers, around 40 million people6
- Due to migration from countries with a high prevalence of β-thalassemia, the disease has spread to other parts of the world6
β-thalassemia is most prevalent in the tropics and subtropical regions (see highlighted areas)6,7

Approximately 200 disease-causing mutations in the β-globin gene have been documented.6 Traditionally, β-thalassemia was classified into 3 main subgroups:8,9
- Thalassemia major:
severe anemia from birth - Thalassemia intermedia:
mild to moderate anemia - Thalassemia minor:
may be borderline symptomatic
Patients are classified as either transfusion dependent or non-transfusion dependent based on the need for transfusions in order to survive5,9
Adapted with permission from Guidelines for the Management of Transfusion Dependent Thalassaemia, 3rd edition, 2014. Reproduction is prohibited.
Current therapy of β-thalassemia is predominantly limited to lifelong red blood cell (RBC) transfusions plus iron chelation therapy (ICT).2,11,12
Red blood cell (RBC) transfusions are an important supportive-care option; however, they are burdensome and carry risks2,5,13-15
- RBC transfusions are a life-saving, supportive-care option in transfusion-dependent β-thalassemia, which has reduced morbidity and improved life expectancy of patients11,12
- RBC transfusions rapidly improve hemoglobin (Hb) levels to compensate for anemia; however, they suppress and lack the ability to correct the underlying ineffective erythropoiesis2,11,12
- The improvement in Hb levels is also temporary, hence regular transfusions are needed in transfusion-dependent β-thalassemia11
- Transfusions are time consuming, taking an average of 3 to 4 hours for treatment, including about 1.9 hours on average per blood unit transfused5,13
- There is a risk of infection from RBC transfusions like hepatitis B and C and, in some populations, HIV.14 Alloimmunization can occur in 10%-20% of patients with β-thalassemia5,16
- Iron overload is the greatest concern associated with regular RBC transfusions4,14
- Because there is no physiological mechanism to excrete excess iron, without chelation, iron overload leads to organ failure and mortality in β-thalassemia5,15
Very few eligible patients receive non-transfusional approaches for managing transfusion-dependent β-thalassemia—such as hematopoietic stem cell transplant or gene therapy.17,18
Due to iron overload, patients with transfusion-dependent
β-thalassemia need to receive iron chelation therapy (ICT)5
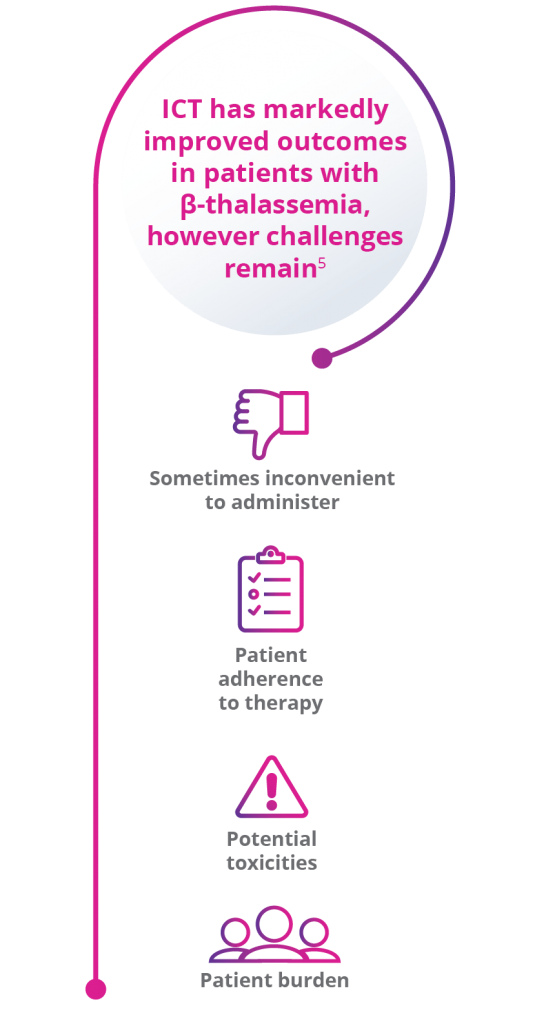
- ICT is used for the management of iron overload, which is an inevitable complication of regular blood transfusions because the body lacks a mechanism to excrete excess iron.5
- Iron chelating agents can have various side effects, such as retinal toxicity, hearing loss, skin rashes, diarrhea, increased liver enzymes, reduced kidney function, arthropathy, or neutropenia2,15
- With the availability of new ICTs and new formulations, adherence has improved but is not yet optimal2,5
There is a significant unmet need for treatments that could reduce RBC transfusions and related burden in adults with β-thalassemia.11,12